

Kidney inflammation in IgAN has its origin in the gut
It has become increasingly apparent in recent decades that the gut is a very versatile multifunctional organ that is not just responsible for the processing of foodstuffs to produce the nutrients required to feed the body. The gut operates in partnership with other organs and can, in certain circumstances, also influence the kidneys. It also acts as an important barrier that protects the body against incursion by pathogens and toxins.1 The mucous membrane of the gut is continually exposed to food antigens and bacteria.2 It is thus important for our health that the immune cells of the intestinal mucous membranes are capable of keeping these potentially threatening agents in check. The gastrointestinal tract has its own immune system for this purpose, a component of which is the gut-associated lymphoid tissue (or GALT). Included in the GALT are Peyer's patches in the small intestine; these are groupings of lymphoid follicles present in the distal ileum (the final section of the small intestine). When these come into contact with antigens, the role of the Peyer’s patches is to induce the formation of IgA1-producing plasma cells. This IgA1 is known as mucosal IgA1 and it exhibits characteristics that differentiate it from other forms of IgA1.3
What is so special about IgA1?
Immunoglobulin A (IgA) generally occurs in the forms of serum IgA and secretory IgA. The IgA in the circulating blood is mainly synthesised in the bone marrow (as part of the endogenous immune system) and has a monomer structure.4 In addition, it is characterised by the presence of galactose residues in the hinge region and undergoes metabolism in the liver.5 Secretory IgA, on the other hand, is in dimer form and is released by the mucous membranes of the respiratory tract and the gut.3 The human IgA antibodies are also divided into two subclasses, IgA1 and IgA2.3, 4
In the case of the aetiology of IgA Nephropathy, it would seem that an important role is played by IgA1 synthesised in Peyer's patches in the mucosa of the small intestine. This special isoform of the IgA1 antibody differs in structural terms from that circulating in the blood serum as the former has a deficiency of galactose residues in the hinge region; it is thus known as galactose-deficient IgA1 (gd-IgA1).3, 4
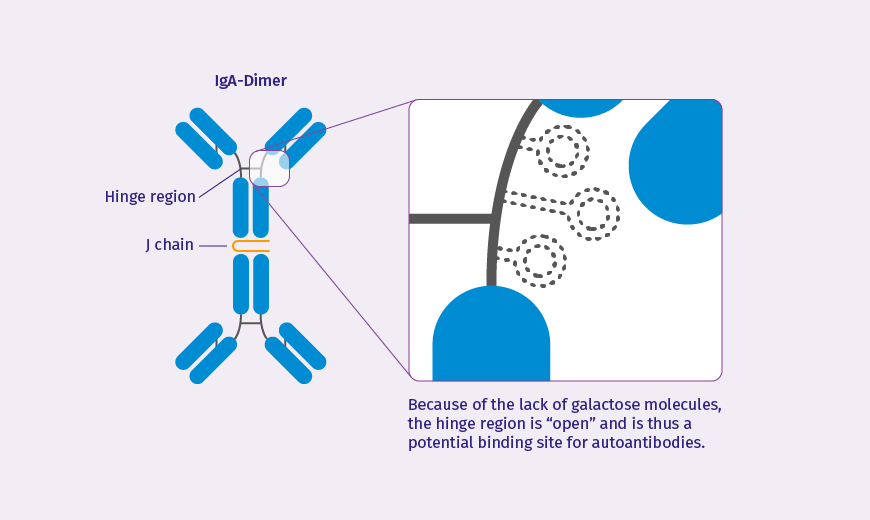
Galactose-deficient IgA1: a complex molecule with a hinge region that is “open” to autoantibodies
Pathogenesis of IgA Nephropathy
In the normal situation, only small quantities of galactose-deficient IgA1 reach the systemic circulatory system.
In individuals with IgAN, however, the opposite is the case: they have raised levels of the galactose-deficient IgA1 that is released from the gut into their systemic circulation. This results in an immune reaction as the galactose deficiency means that this immunoglobulin is identified as an exogenous agent.6 Because of this, IgA and IgG autoantibodies are activated that bind to the open hinge region of the IgA1 molecules. The result is pathogenic immune complexes that begin to accumulate in the glomerular mesangium of the kidneys. These complexes can subsequently lead to activation of the complement system resulting in kidney inflammation and damage, ultimately in deterioration of renal function.3, 4, 6 Hence, galactose-deficient IgA1, that in the physiological situation should not be present in the circulating blood, can be the primary cause of IgA Nephropathy.
In the light of this insight into the aetiology of IgA Nephropathy, it is possible to develop innovative treatment approaches that target the original cause.7, 8
The pathogenesis of IgA Nephropathy
Peyer's patches

Peyer's patches
Peyer's patches in the mucous membrane of the distal ileum synthesise IgA.
Galactose-deficient IgA1

Galactose-deficient IgA1
Among this IgA is galactose-deficient IgA1 with an open hinge region.
Identification by the immune system
![Illustration showing galactose-deficient IgA1 in the circulating blood]](https://06.cdn.stada.com/media/pzrlzylb/graph-pathophys-verlauf-3-en.png?format=webp&width=690&height=690&quality=85)
Identification by the immune system
If galactose-deficient IgA1 reaches the bloodstream, it is identified as an exogenous agent by the immune system and autoantibodies bind to it.
IgA Nephropathy

IgA Nephropathy
The resultant pathogenic immune complexes accumulate in the glomerular mesangium, which can eventually result in the development of IgA Nephropathy.

The high rate of recurrence even after receiving a kidney transplant is evidence that there is an extrarenal cause of IgA Nephropathy.
The assumption that this Nephropathy has an extrarenal trigger is confirmed by the fact that there is a high rate of recurrence following what is considered successful kidney transplant surgery.
In the past, patients with end-stage renal disease were given priority for kidney transplants. However, recurrence occurs in some 33% of kidney transplant patients who were affected by IgA Nephropathy. Onset can be directly after transplantation, although on average it is first diagnosed 3 years later.9 It is particularly the case that if there is significant elevation of IgA serum levels within 12 months of surgery, there is an increased risk of the recurrence of IgA Nephropathy.10


